Multiple sequence alignment
Last updated on 2026-03-31 | Edit this page
Overview
Questions
- How do you align multiple sequences?
- Why is it important to properly align sequences?
Objectives
- Use
mafftto align multiple sequences. - Test different algorithms.
Introduction
In this episode, we are exploring multiple sequence alignment (MSA).
In the first task, you are going to use mafft
to align homologs of RpoB, the β subunit of the
bacterial RNA polymerase. It is a long, multi-domain protein, suitable
for showing issues related to MSA.
In the second task, you will trim that alignment to remove poorly aligned regions.
But first, go to your own folder and create a phylogenetics subfolder. You will use the alignments for other tutorials as well.
Task 1: Use different flavors of MAFFT and compare the
results
Start by finding the data, which is a fasta file containing 19 sequences of RpoB,
gathered by aligning RpoB from E. coli to a very small database
present at NCBI, the landmark
database. These sequences are a subset of the sequences gathered in the
previous exercise on BLAST.
The file is available in
/proj/g2020004/nobackup/3MK013/data/.
Challenge 1.1: prepare the terrain
The course base folder is at
/proj/g2020004/nobackup/3MK013. Go to your own folder,
create a phylogenetics subfolder, and move into it. Also,
start the interactive session, for 4 hours. The session
name is uppmax2025-3-4 and the cluster is
snowy.
Challenge 1.2: copy the file
Copy the file to your newly created phylogenetics
folder. Use a relative path.
Use the ../../ to see what’s two folders up, and then
data/rpoB/rpoB.fasta
Challenge 1.1: prepare the terrain (continued)
Renaming sequences
Look at the accession ids of the fasta sequences: they are not very informative.
OUTPUT
>WP_000263098.1 MULTISPECIES: DNA-directed RNA polymerase subunit beta [Enterobacteriaceae]
>WP_011070610.1 DNA-directed RNA polymerase subunit beta [Shewanella oneidensis]
>WP_003114335.1 DNA-directed RNA polymerase subunit beta [Pseudomonas aeruginosa]
>WP_002228870.1 DNA-directed RNA polymerase subunit beta [Neisseria meningitidis]
>WP_003436174.1 MULTISPECIES: DNA-directed RNA polymerase subunit beta [Eubacteriales]Keep the taxonomic name following the accession id, separated by
_, using a bit of sed magic. Save the
resulting file for later use, and show the headers again.
BASH
sed "/^>/ s/>\([^ ]*\) \([^\[]*\)\[\(.*\)\]/>\\3_\\1/" rpoB.fasta | sed "s/ /_/g" > rpoB_renamed.fasta
grep '>' rpoB_renamed.fasta | head -n 5Nerd alert: dissecting the sed
magic
This is optional reading.
The sed
command matches (and possibly substitutes) strings (chains of
characters). In that case, the goal is to simplify the header by putting
the taxonomic group first but keeping it informative enough by adding
the accession number. The strategy is to match what is between the
> and the first space, then what is between square
parentheses, and put them back, separated by an underscore.
The sed command first matches only lines that start with
a > (/^>/). It then substitutes (general
pattern s/<something>/<something else>/) a text
with another one. The first part is to match the accession id, between
(escaped) square brackets, which comes after the > at
the beginning of the line. This is expressed as
>\([^ ]*\): match any number of non-space characters
([^ ]*) and put it in memory (what is between
\( and \)). Then, the description is matched
by \([^\[]*\), any number of characters that are not an
opening bracket [, and put into memory. Finally, the
taxonomic description is matched: \[\(.*\)\], that is, any
number of characters between square brackets is stored into memory. The
whole line is then replaced with a >, the third match
into memory, followed by an _ and the content of the first
match into memory >\\3_\\1. Then, all the input is
passed through sed again, to replace any space with an underscore:
s/ /_/g and the output is stored in a different file.
OUTPUT
>Enterobacteriaceae_WP_000263098.1
>Shewanella_oneidensis_WP_011070610.1
>Pseudomonas_aeruginosa_WP_003114335.1
>Neisseria_meningitidis_WP_002228870.1
>Eubacteriales_WP_003436174.1It looks better.
Alignment with progressive algorithm
Use mafft with a progressive algorithm to align the
sequences.
Challenge 1.3: Run mafft with
progressive algorithm
Use the FFT-NS-2 algorithm from mafft to align the
renamed sequences. Also, record the time it takes for mafft
to complete the task.
Use the module command to load
bioinfo-tools and MAFFT. Use time
to record the time.
The help obtained through mafft -h is not very
informative about algorithms, so check the mafft
webpage.
mafft actually has one executable program for each
algorithm, all starting with mafft-. A way to display them
all is to type that and push the Tab key twice to see all
possibilities.
BASH
module load <general module> <mafft module>
time mafft-<algorithm> <fasta_file> > rpoB.fftns.alnOUTPUT
...
[...]
Strategy:
FFT-NS-2 (Fast but rough)
Progressive method (guide trees were built 2 times.)
If unsure which option to use, try 'mafft --auto input > output'.
For more information, see 'mafft --help', 'mafft --man' and the mafft page.
The default gap scoring scheme has been changed in version 7.110 (2013 Oct).
It tends to insert more gaps into gap-rich regions than previous versions.
To disable this change, add the --leavegappyregion option.
real 0m1.125s
user 0m0.818s
sys 0m0.181sThe last line is the output of the time command. It took
1.125 seconds to complete this time.
Type mafft and try tab-complete to see all versions of
mafft.
Try the command time
Alignment with iterative algorithm
Now use one of the supposedly better iterative algorithm of
mafft to align the same sequences. Choose the E-INS-i
algorithm which is suited for proteins that have highly conserved
motifs interspersed with less conserved ones.
Take a few minutes to read upon the different alignment strategies on the page above.
Challenge 1.4
Use the superior E-INS-i algorithm from mafft to align the renamed
sequences. Also, record the time it takes for mafft to
complete the task.
OUTPUT
[...]
Strategy:
E-INS-i (Suitable for sequences with long unalignable regions, very slow)
Iterative refinement method (<16) with LOCAL pairwise alignment with generalized affine gap costs (Altschul 1998)
If unsure which option to use, try 'mafft --auto input > output'.
For more information, see 'mafft --help', 'mafft --man' and the mafft page.
The default gap scoring scheme has been changed in version 7.110 (2013 Oct).
It tends to insert more gaps into gap-rich regions than previous versions.
To disable this change, add the --leavegappyregion option.
Parameters for the E-INS-i option have been changed in version 7.243 (2015 Jun).
To switch to the old parameters, use --oldgenafpair, instead of --genafpair.
real 0m7.367s
user 0m7.022s
sys 0m0.244sIt now took 7.36 seconds to complete this time, i.e. 6 times more than with the progressive algorithm. It doesn’t make a big difference now, but with hundreds of sequences it will make one.
Alignment visualization
You will now inspect the two resulting alignment methods. There are
no convenient way to do that from Uppmax, and the easiest solution is to
download the alignments on your computer and to use
either seaview
(you will need to install it on your computer) or an online alignment
viewer like AlignmentViewer.
Arrange the two windows on top of each other. Change the fontsize (Props -> Fontsize in Seaview) to 8 to see a larger portion of the alignment.
Challenge
Can you spot differences? Which alignment is longer?
Hint: try to scroll to position 800-900. What do you see there? How are the blocks arranged?
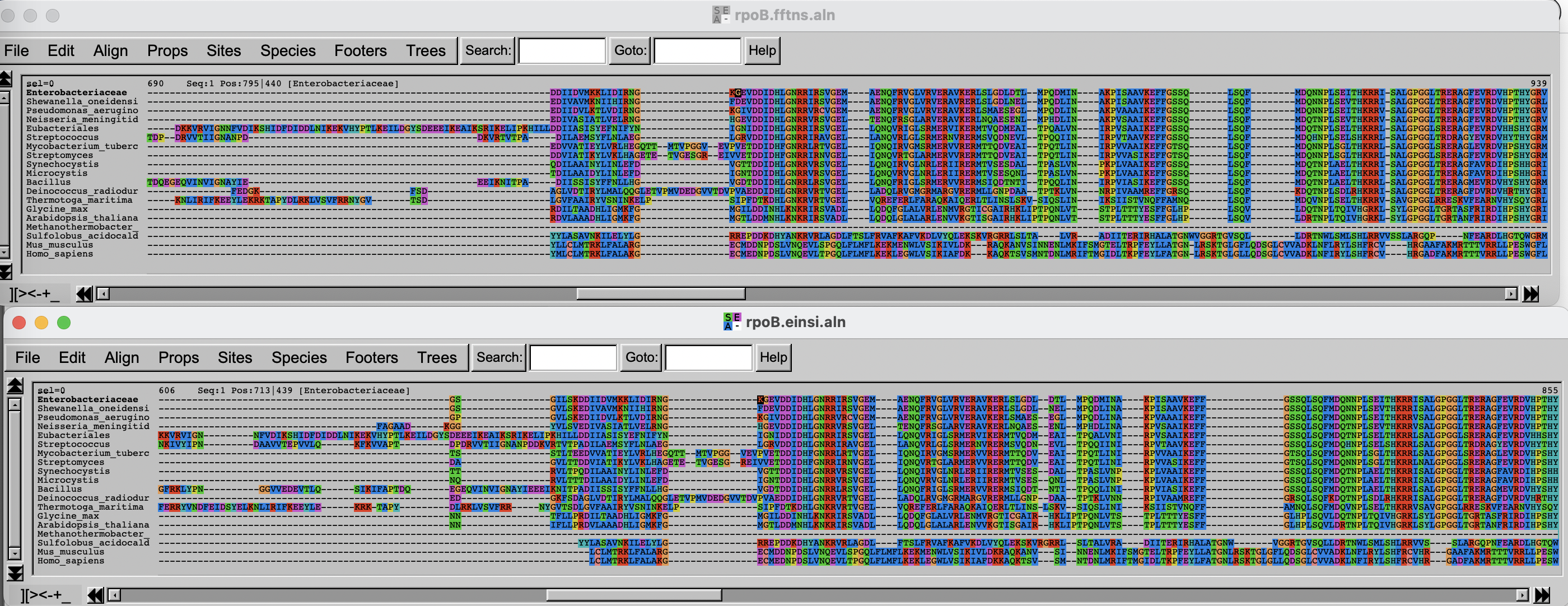
If you have time over, spend it exploring the different options of Seaview/AlignmentViewer.
Task 2: Trim the alignment
Often, some regions of the alignment don’t align properly, either because they contain low complexity segments (hinges in proteins) or evolved through repeated insertions/deletions, which alignment program cannot handle properly. It is thus good practice to remove (trim) these regions, as they are likely to worsen the quality of the subsequent phylogenetic trees. On the other hand, trimming too much of the alignment removes also potentially valuable information. There is thus a balance to be found.
In this part, you will use two different alignment trimmers, TrimAl and ClipKIT, on the
results of mafft’s E-INS-i algorithm.
Trimming with TrimAl
First, load the module and look at the list of options available with
trimAl.
trimAl offers a lot of different options. You are going
to explore two different: gap threshold (-gt) and the
heuristic method -automated1, which automatically decides
between three automated methods, -gappyout,
-strict and -strictplus, based on the
properties of the alignment. The gap threshold methods removes columns
that contain a fraction of sequences lower than the cut-off.
For comparison purposes, you will be adding an html output.
Challenge 2.1: TrimAl with gap threshold
Use trimAl to remove positions in the alignment that
have more than 40% gaps.
The “gap threshold” is actually expressed as fraction of non-gap residues.
Challenge 2.2: TrimAl with automated trimming
Use trimAl with the automated heuristic algorithm.
Add a -automated1 option and rerun as above, choosing a
different output file.
Challenge 2.3: Compare the results
Get the files (both alignments and html files) to your own laptop and
visualize the results, by opening the html files with your
browser and the alignment files with seaview or the viewer
you used above.
On your own laptop, go inside the folder where you want to import the
files. Use scp. On some OS it is necessary to escape the
wildcard *. If the output says something about
no matches found, try that.
As before, if you do not have access to a terminal on your windows laptop, use MobaXterm and Session > SFTP to copy files to your computer.
Trimming with ClipKIT
ClipKIT is
one of the more recent tools to trim multiple sequence alignments. In a
nutshell, it tries to preseve phylogenetically-informative sites, rather
than trimming gappy regions. Although it also has multiple options and
modes, you will only use the default mode, smart-gap.
OUTPUT
---------------------
| Output Statistics |
---------------------
Original length: 2043
Number of sites kept: 1543
Number of sites trimmed: 500
Percentage of alignment trimmed: 24.474%
Execution time: 0.379sComparing all the results
Import the data and inspect the three alignments.
Challenge 2.5: Import data and compare results
Copy the alignment file and visualize with seaview or
the web-based visualization tool.
Use scp as above.
Then use seaview or another viewer to visualize and
compare results.
The three alignments on top of each other look like this. Click on this link to better see the figure.
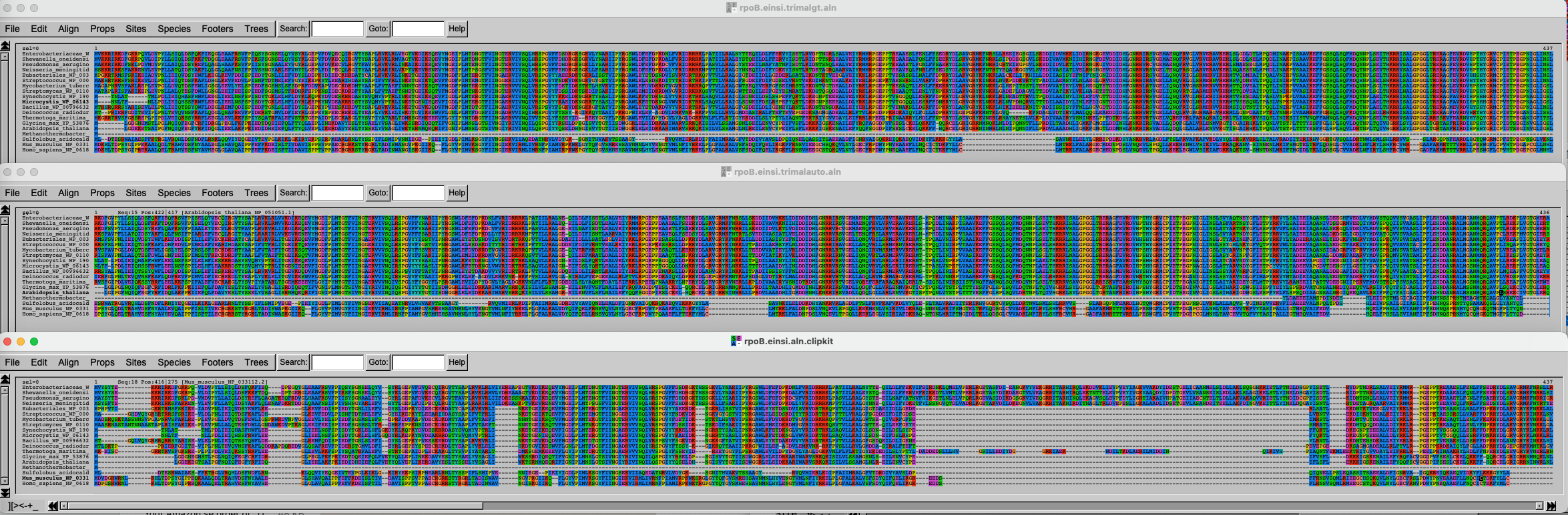
It is of course difficult to draw conclusions based on this figure, but can you spot some trends? What alignment is more likely to generate good results?