Mapping
Last updated on 2026-04-01 | Edit this page
In this episode we will try to pinpoint single nucleotide polymorphism (SNPs), also called single nucleotide variants (SNVs), between our samples and the reference. The SNPs are determined by a process called read mapping in which reads are aligned to the reference sequence.
Overview
Questions
- How to generate a phylogenetic tree from SNP data?
Objectives
- Map reads against a reference genome
- Extract single nucleotide polymorphisms
- Establish a phylogenetic tree from the SNP data

The identified SNPs will be used to compare the isolates to each other and to establish a phylogenetic tree.
SNP calling
snippy is a pipeline that contains different tools to determine SNPs in sequencing reads against a reference genome. It takes forward and reverse reads of paired-end sequences and aligns them against a reference.
Challenge
First we’ll start the interactive session, load the snippy module and
create a snps folder in the molepi/results to
hold the results from snippy.
Snippy is fast but a single run still takes about 15 minutes. We would therefore tell snippy to run all the samples after each other. However this time we cannot use a wildcard to do so. We would instead run all the samples in a loop. The “real” loop is commented out, and we use one that runs only the sample we’ve analyzed so far.
BASH
cd ../data/trimmed_fastq/
#for sample in ERR026473 ERR026474 ERR026478 ERR026481 ERR026482 ERR029206 ERR029207
for sample in ERR029206
do
snippy --ram 8 --outdir ../../results/snps/"${sample}" --ref ../GCF_000195955.2_ASM19595v2_genomic.fna --R1 "${sample}"_1.fastq.gz_trim.fastq --R2 "${sample}"_2.fastq.gz_trim.fastq
doneHere, we provide snippy with an output folder
(--outdir), the location of the reference genome
(--ref), and the trimmed read files for each end of the
pair (--R1 and --R2). We also indicate that
snippy should use 8 Gb of memory (--ram 8).
OUTPUT
CHROM POS TYPE REF ALT EVIDENCE FTYPE STRAND NT_POS AA_POS EFFECT LOCUS_TAG GENE PRODUCT
NC_000962.3 1849 snp C A A:217 C:0
NC_000962.3 1977 snp A G G:118 A:0
NC_000962.3 4013 snp T C C:176 T:0
NC_000962.3 7362 snp G C C:156 G:0
NC_000962.3 7585 snp G C C:143 G:0
NC_000962.3 9304 snp G A A:127 G:0
NC_000962.3 11820 snp C G G:164 C:0
NC_000962.3 11879 snp A G G:125 A:0
NC_000962.3 14785 snp T C C:163 T:1This list gives us information on every SNP that was found by snippy when compared to the reference genome. The first SNP is found at the position 1849 of the reference genome, and is a C in the H37Rv (reference strain) and an A in isolate ERR029206. There is a high confidence associated with this SNP: an A has been found 217 times in the sequencing reads and never a C at this position.
Now copy a reduced version of the results obtained from the other
samples, save it in the results folder. We will also rename
the existing snps folder to save it. We extract them from
the archive in the data folder, using tar.
tar is a program to take multiple files, including their
folder structure and put them in a single tar “ball”. Often
the tarballs are compressed with gzip.
BASH
cd /proj/g2020004/nobackup/3MK013/<username>/molepi/results
mv snps snps_partial
mkdir snps
cd snps
tar xvzf ../../../../data/snps.tar.gz
ls *OUTPUT
ERR026473:
ref.fa. snps.bam.bai snps.filt.vcf snps.log snps.tab snps.vcf.gz
ref.fa.fai snps.bed snps.gff snps.raw.vcf snps.txt snps.vcf.gz.csi
snps.aligned.fa snps.csv snps.html snps.subs.vcf snps.vcf
ERR026474:
ref.fa snps.bam.bai snps.filt.vcf snps.log snps.tab snps.vcf.gz
ref.fa.fai snps.bed snps.gff snps.raw.vcf snps.txt snps.vcf.gz.csi
snps.aligned.fa snps.csv snps.html snps.subs.vcf snps.vcfNote the unusual way to pass arguments to tar with just
xvzf instead of -x -v -z -f or
-xvzf. x tells tar to extract the archive
(c would be used to create the archive); v is
for verbose, providing more information on the output; z
tells tar to decompress the file; f tells what file to work
on.
The folders don’t contain everything that is output by
snippy, to save space. But it is enough to run
snippy-core, which summarizes several snippy
runs.
Challenge: How many SNPs were identified in each sample??
Find out how many SNPs were identified in the M.
tuberculosis isolates when compared to H37Rv. Hint: The
.txt file in the snippy output contains summary
information:
Challenge: How many SNPs were identified in each sample?? (continued)
OUTPUT
DateTime 2023-05-17T13:51:57
ReadFiles [...]/data/trimmed_fastq/ERR029207_1.fastq.gz_trim.fastq [...]/data/trimmed_fastq/ERR029207_2.fastq.gz_trim.fastq
Reference [...]/data/GCF_000195955.2_ASM19595v2_genomic.fna
ReferenceSize 4411532
Software snippy 4.6.0
Variant-COMPLEX 32
Variant-DEL 57
Variant-INS 52
Variant-MNP 2
Variant-SNP 1290
VariantTotal 1433This M. tuberculosis isolate contains 1290 SNPs compared to H37Rv.
Repeat this for the other isolates. How about using a
for loop?
Core SNPs
In order to compare the identified SNPs with each other we need to know if a certain position exists in all isolates. A core site can have the same nucleotide in every sample (monomorphic) or some samples can be different (polymorphic).
In this second part, after identifying them, snippy will concatenate the core SNPs, i.e. ignoring sites that are monomorphic in all isolates and in the reference. Concatenation of the SNP sites considerably reduces the size of the alignment.
The --ref argument provides the reference genome. Each
folder containing the result of the previous step of snippy
is then added to the command line.
BASH
cd /proj/g2020004/nobackup/3MK013/<username>/molepi/results/snps
snippy-core --ref=../../data/GCF_000195955.2_ASM19595v2_genomic.fna ERR026473 ERR026474 ERR026478 ERR026481 ERR026482 ERR029206 ERR029207Instead of writing all samples explicitly, we could use wildcards:
BASH
cd /proj/g2020004/nobackup/3MK013/<username>/molepi/results/snps
snippy-core --ref=../../data/GCF_000195955.2_ASM19595v2_genomic.fna ERR*The last few lines look like this:
OUTPUT
...
Loaded 1 sequences totalling 4411532 bp.
Will mask 0 regions totalling 0 bp ~ 0.00%
0 ERR026473 snp=1378 del=174 ins=165 het=747 unaligned=125683
1 ERR026474 snp=1369 del=180 ins=143 het=811 unaligned=123020
2 ERR026478 snp=1381 del=221 ins=143 het=638 unaligned=112145
3 ERR026481 snp=1349 del=215 ins=138 het=754 unaligned=126217
4 ERR026482 snp=1348 del=215 ins=145 het=911 unaligned=127554
5 ERR029206 snp=1352 del=152 ins=98 het=1839 unaligned=122677
6 ERR029207 snp=1355 del=152 ins=97 het=1900 unaligned=118570
Opening: core.tab
Opening: core.vcf
Processing contig: NC_000962.3
Generating core.full.aln
Creating TSV file: core.txt
Running: snp-sites -c -o core.aln core.full.aln
This analysis is totally hard-core!
Done.Our output was written to ‘core.aln’. But let’s have a look at the results.
Discussion: What’s in the output of snippy??
Have a look at the content of these three files>
core.aln
core.full.aln
core.tab
core.txt
What is the difference between these files? Why is
core.aln smaller than core.full.aln? What is
in core.aln?
Phylogenetic tree
We will here infer a phylogenetic tree from the file ‘core.aln’ with IQ-TREE.
In this case, we want IQ-TREE to automatically select the best
(nucleotide) substitution model. IQ-TREE does that by testing many
(among a very large collection) substitution models. We also have SNP
data, which by definition do not contain constant (invariable) sites. We
thus input the alignment with the -s option and the model
with -m MFP+ASC. MFP will tell IQ-TREE to test
a range of models, and ASC will correct for the fact that
there is no constant sites.
Challenge
Infer a phylogenetic tree with IQ-TREE, using the aligned polymorphic
sites identified by snippy-core above. IQ-TREE should find
the best possible model, adding the correction for no constant sites.
Don’t forget to load the right module.
OUTPUT
...
Total wall-clock time used: 0.312 sec (0h:0m:0s)
Analysis results written to:
IQ-TREE report: core.aln.iqtree
Maximum-likelihood tree: core.aln.treefile
Likelihood distances: core.aln.mldist
Screen log file: core.aln.log
Date and Time: Thu May 18 09:00:37 2023With this small data set, IQ-TREE finishes very quickly. Let’s put the resulting files into a separate folder and let’s rename our resulting tree.
Let’s inspect our tree, and give it another extension to make it clear it is a newick file.
OUTPUT
(ERR026473:0.0004854701,ERR026474:0.0004356437,((((ERR026478:0.0000010000,ERR029207:0.0000010000):0.0000010000,ERR029206:0.0000131789):0.0006712219,Reference:0.0075887710):0.0000152316,(ERR026481:0.0000066238,ERR026482:0.0000131899):0.0005602421):0.0002373720);This does not look much like a tree yet. The tree is written in a bracket annotation, the Newick format. In order to make sense of it we can better view this in a tree viewer.
Challenge: Can you identify what substitution model IQ-TREE used?
What model was selected? How does IQ-TREE choose between different models? What does e.g. “JC+ASC+G4” means?
The log file of IQ-TREE contains a lot of information.
The IQ-TREE website contains a description of the DNA substitution models.
OUTPUT
...
Best-fit model according to BIC: TVMe+ASC
...IQ-TREE chose the TVMe+ASC model. We’ve discussed the ASC above, and you can read more about the TVMe in the IQ-TREE manual. In short, it is a mdoel where base frequencies are deemed equal, and all rates estimated independently, except the rates of transversions (A <-> G and C <-> T) are equal.
The model selection process is also described on the IQ-TREE website.
Challenge: Visualize the tree on your computer
Copy the tree in Newick format (core.aln.newick) to your
computer using scp and use FigTree or phylo.io to visualize it. Do you see a way
to assess the quality of the tree?
Challenge: Rerun the tree, calculating bootstraps this time
In the Phylogenetics module, we
calculated 1000 UltraFast bootstraps using the -B 1000
option. Here, calculate 100 non-parametric bootstraps. Non-parametric
bootstraps are bootstraps that are calculated without making assumptions
about the data. In that case, it means actually resampling the alignment
and calculating trees for each of the bootstrapped alignments. It is a
much slower process than the UltraFast method (which is parametric), but
in that case, since the alignment is very small, the computational load
is acceptable.
With IQ-TREE, non-parametric
bootstraps are inferred using the
-b <n bootstraps> option. Make sure all files are
prefixed with core.aln.100npb (option
--prefix).
The process will take a minute or so. Then in the
results folder, next to the tree folder,
create a tree100npb folder and move the results file
there.
Challenge: Can you identify what substitution model IQ-TREE used? (continued)
When that finishes, copy the resulting tree to your local computer
and visualize it. Display support values: - With FigTree: on the left
menu, tick “Branch labels”, and choose the last row (what you entered
when prompted to do so, label by default) - With phylo.io:
when loading the tree, tick the “Use internal label for branches”. Then
click on “Settings” > “Branches & Labels”, use the drop menu “A”
under “Node labels” and choose “Data”.
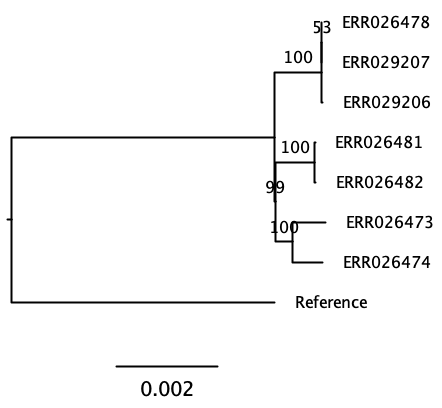
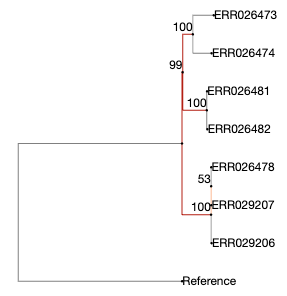
As shown by phylo.io, beta version. Note the position of the “99”.
- Single nucleotide polymorphisms can be identified by mapping reads to a reference genome
- Parameters for the analysis have to be selected based on expected outcomes for this organism
- Concatenation of SNPs helps to reduce analysis volume
- Phylogenetic trees can be written with a bracket syntax in Newick format